Tot l’anàlisi d’aquest treball s’ha realitzat amb el sistema operatiu Unix.
Totes les proteïnes (incloent selenoproteïnes, homòlegs amb cisteïna o altres aminoàcids i maquinària de biosíntesi) han estat analitzades pels diferents membres del grup, comparant el nostre genoma (Ailuropoda melanoleuca) amb les proteïnes de Mus musculus i Homo sapiens, presents a la base de dades SelenoDB, ja que filogenèticament són de les espècies amb selenoproteaoma identificat més properes a l’ós panda.
Els resultats presentats a la pàgina web fan referència a una de les dues espècies, en cada cas aquella que hem considerat més adequada per comparar amb el nostre genoma, a partir dels resultats del tBlastn i del Selenoprofiles.
A més hem utilitzat altres programes com el tRNAscan-SE i Jalview per aportar informació adicional útil per interpretar els resultats.
Per acabar de contrastar els nostres resultats, hem utilitzat el programa Selenoprofiles per corroborar els resultats obtinguts.
S’han realitzat les mateixes comparacions a partir de programes automatitzats fets per nosaltres (Automatització).
1. Seqüències de les Selenoproteïnes
Totes les seqüències de les selenoproteïnes utilitzades per l’anàlisi del selenoproteoma de l'ós panda han estat extretes de la bases de dades SelenoDB. Tot i així, cal tenir en compte que a SelenoDB no hi ha totes les selenoproteïnes identificades en les diferents espècies i per aquesta raó, hem hagut de recórrer a la cerca en altres bases de dades com l'Ensmbl o l'NCBI per acabar de completar el selenoproteoma d'Ailuropoda melanoleuca.
Les seqüències aminoacídiques emprades han estat les d’H. sapiens i M. musculus.
I les podeu trobar a la pàgina web de SelenoDB.
2. El genoma d'Ailuropoda melanoleuca
El genoma d’Ailuropoda melanoleuca ha estat proporcionat pels professors de l’assignatura. A partir del següent path al terminal/shell:
3. Programari informàtic utilitzat
BLAST (Basic Local Alignment Search Tool),és un programa informàtic que alinea localment una seqüència problema, o query, amb seqüències provinents d’una base de dades. Per tal de realitzar l’alineament, el programa segueix un algoritme heurístic que selecciona ràpidament les seqüències (hits) amb més homologia respecte la seqüència problema.
Aquestes seqüències poden ser de naturalesa aminoacídica o nucleotídica, condicionant el tipus de BLAST que serà utilitzat. En el nostre estudi interessa comparar una seqüència de proteïna extreta de SelenoDB amb el genoma del panda. Per tal de comparar proteïna amb la base de dades de nucleòtids s’utilitza el tBLASTn, que tradueix totes les seqüències nucleotídiques en els 6 possibles ORFs (Open Reading Frames o marcs de lectura) per tal de comparar-les amb la seqüència problema i finalment mostrar les homologies trobades a partir de hits.
Extracció del software de tBLASTn:
Cal introduir dues ordres al terminal/shell que indiquen els paths necessaris per a que el programa es pugui executar:
Execució del tBLASTn: Cal introduir la comanda següent:
Llegenda. –p: tipus de blast, -i: ubicació de la query, -d: genoma del panda i –o: output.
L’output del tBLASTn és un llistat dels hits trobats. Aquests indiquen les regions del genoma que tenen alta similitud amb la seqüència probema/query.
La significança es pot valorar amb l’e-value present a cada hit. Aquest paràmetre descriu el nombre de hits que es poden esperar únicament per atzar quan realitzem l'anàlisi amb BLAST, i és significatiu quan és equivalent o inferior a 10-4. D’aquesta manera, sempre s’escollirà el hit amb e-value més petit.
Afegint l'opció -m9 s'organitzen els resultats del BLAST en forma de taula, de manera que es poden veure les posicions que ocupen els alineaments. Amb el que es poden determinar les posicions que ocupen les seqüències d’interès dins el cromosoma (això será important per realizar el Fastafetch).
Aquest programa requereix l’extracció prèvia de regions genòmiques amb alta potencialitat de ser selenoproteïnes.
Per tal d’utilitzar els següents programes cal introduir al terminal/shell la següent comanda:
L’exonerate requereix tot un seguit de comendes prèvies per tal d’extreure les seqüències que interessen del genoma del panda. A partir de les dades obtingudes al tBLASTn es pot determinar les posicions i mides d’aquestes seqüències.
1. Fastaindex
Organitza el fitxer del genoma en diferents regions, és a dir organitza fitxers multifasta a forma indexada per tal de poder extreure les parts a analitzar amb el programa Fastafetch .
Llegenda. S’indica primer la ubicació del genoma i després la ubicació del fitxer de sortida.
2. Fastafetch
A partir del document sortida.index, selecciona una regió indicada i la desa en un document fasta.
Llegenda:Primer s'indica la ubicació del genoma.
nomseq: nom de la regió que volem extreure (la que té l’e-value inferior) i nomseq.fa: fitxer de sortida.
3. Fastasubsequence
Selecciona una zona encara més delimitada de la regió, concretament la que envolta el gen que estem buscant. D’aquesta manera, obtenim una seqüència més curta, amb la que serà més fàcil treballar. S’ha d’agafar les posicions del hit obtingut al BLAST i expandir els marges de manera que ens assegurem la presència del gen upstream i downstream.
S’indica primer la ubicació del genoma.
Llegenda. start: inici de la subseqüència (aa), lengthnúmero de nucleòtids que volem extreure i genomic.fa:fitxer de sortida.
La longitud i posició d’aquestes regions es poden estimar a partir de les dades de longitud dels hits resultants del tBLASTn. En el nostre cas s’han afegit uns 10.000 nucleòtids banda i banda de la regió sel·lecionada per tal d’abarcar el màxim de seqüència evitant així perdre el codó UGA.
4. Exonerate
L’Exonerate és un programa que fa alineament de seqüències d’una manera més exacta que el BLAST, i també més informativa ja que indica més característiques del gen (introns, exons, zones d’splicing, etc).
Llegenda. -m p2g és el model d’alineament (proteïna vs genoma), --showtargetgff: mostrar fitxer de sortida en format GFF, -q: "query", -t indica la subseqüència delimitada anteriorment i sortida.gff fitxer de sortida en format gff.
Exonerate inclou dos programes que seran els passos finals per extreure la seqüència: FastaseqfromGFF.pl i el Fastatranslate:
4.1. FastaseqfromGFF.pl
Per tal d’extreure la seqüència exònica en format fasta a partir de l’arxiu sortida.gff, fem servir el FastaseqfromGFF.pl, programa perl que construeix una sequència de nucleòtids a partir de d’un fitxer GFF. Com que volem construir la seqüència present en els exons (el cDNA), farem servir aquesta comanda per introduir al programa només els exons:
Llegenda: egrep: selecciona les línies on aparegui el patró definit, -w: patró és una paraula sencera, exon:patró que volem buscar, sortida.gff fitxer objectiu i cDNA.gff: fitxer de sortida.
Ja podem fer servir el programa fastaseqfromGFF.pl amb la següent comanda:
Llegenda: genomic.fa subseqüència extreta anteriorment, cDNA.gff arxiu en format gff contenint només els exons i cDNA.fa fitxer de sortida que contindra en format fasta la seqüència de DNA del cDNA.
4.2. Fastatranslate
Aquest pas permetrà traduir el cDNA obtingut a seqüència d’aminoàcids, resultant en un fitxer de sortida que inclou els sis ORFs possibles. D’aquests sis ORFs cal escollir-ne un, normalment el de del patró de lectura 1 o -1.
Llegenda: cDNA.fa és la ubicació del cDNA i aa_gen.mfa és el fitxer de sortida en format multifasta.
El programa Genewise genera una nova anotació del gen, a l’igual que l’Exonerate també descriu exons, introns i zones d’splicing. Ara bé, l’algorisme que utilitza no és el mateix, de manera que pot ajudar a contrastar la informació obtinguda amb la resta de programari.
Per tal de fer-lo anar, executem la següent comanda:
Llegenda: -pep: mostrar al fitxer de sortida la seqüència peptídica predita,-pretty: mostrar l’alineament, -cdna mostrar la seqüència genòmica alineada,-gff: resultat en format gff, query.fa és la ubicació de la nostra query, genomic.fa és la subseqüència i sortida.gff és el fitxer de sortida.
El T-Coffee és un programa que permet realitzar alineaments múltiples de proteïna, DNA i RNA.
D’aquesta manera, a partir de la seqüència aminoacídica sense introns el T-Coffe realitza un alineament global de la proteïna resultant amb la seqüència de la proteïna problema extreta del SelenoDB (query). El resultat indicarà presència o absència d’homologia entre les dues seqüències.
Llegenda. fitxerFASTAsequencia1: la "query" i fitxerFASTAsequencia2: proteïna obtinguda amb l’exonerate o el genewise, o viceversa.
Els últims passos per determinar si una seqüència correspon a una selenoproteïna és la búsqueda d'elements SECIS i de maquinària de traducció de la proteïna.
Per tal de detectar els elements SECIS de la seqüència cal accedir al programa online< a href="http://genomics.unl.edu/SECISearch.html"> SECISearch. Aquest programa també es pot executar al terminal/Shell, però cal exportar-lo:
Per executar el programa utilitzarem la comanda següent:
Llegenda. genomic.fa: seqüència problema.
El tRNAscan-SE és un programa que busca els tRNAs necessaris per a la traducció d’aquella proteïna. Hem instal·lat el programa tRNAscan i posteriorment, hem passat el nostre genoma pel programa.
S'ha obtingut un fitxer amb tots els possibles tRNAs del genoma d'Ailuropoda melanoleuca, a partir d'això hem realitzat la següent comanda per tal de seleccionar només aquells tRNAs específics de selenocisteïna:
Finalment, després d'un anàlisi exhaustiu considerem que els tRNAs que presenten un cove score més elevat seran els més probables. Així doncs, podem dir que l'ailuropoda melanoleuca presenta dos tRNAs .(link amb totes les sec i marcades amb coloret).
A continuació mostrem la estructura d'aquests:
GL192530.1.trna29 (930778-930848) Length:71 bp Type: SeC Anticodon: TCA at 33-35 (930810-930812) Score: 50.79 Seq: ACCTGGCTGGCTCAGTCGGTAGAGCATGTGACTCAATCTCATGGtTGTGAGTTCGAGACCCACGTTGGGTA Str: >>>>>>>..>>>>........<<<<.>>.>>.....<<.<<.....>>>.>.......<.<<<<<<<<<<.
Per veure la imatge clica aquí.
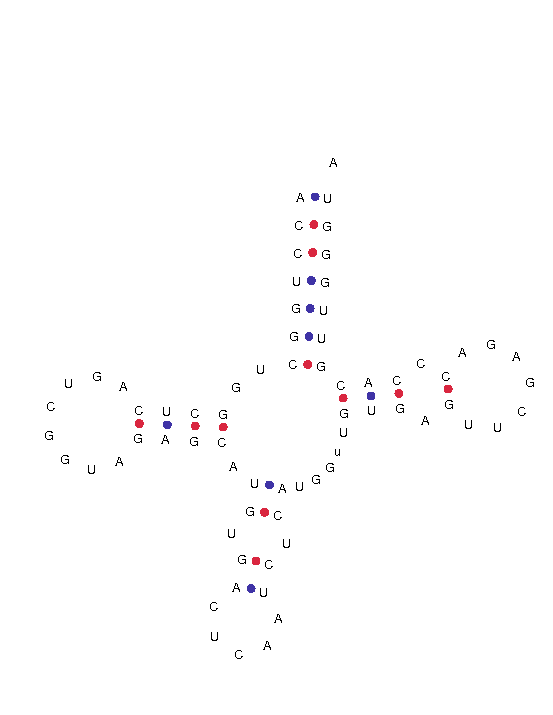{kind=link}
GL193124.1.trna3 (26915-27000) Length: 86 bp Type: SeC(e) Anticodon: TCA at 36-38 (26950-26952) Score: 73.19 Seq: GCCCGAATGATCCTCAGTGGTCTGGGGTGCAGGCTTCAAACCTGTAGCTGTCTAGCGACAGAGTGGTTCAATTCCACCTTTCGGGC Str: >>>>>>>.>..>>>>>>....<<<<<<>>>>>>.......<<<<<<.>>>>>....<<<<<.>>>>.......<<<<<.<<<<<<<
Per veure la imatge clica aquí.
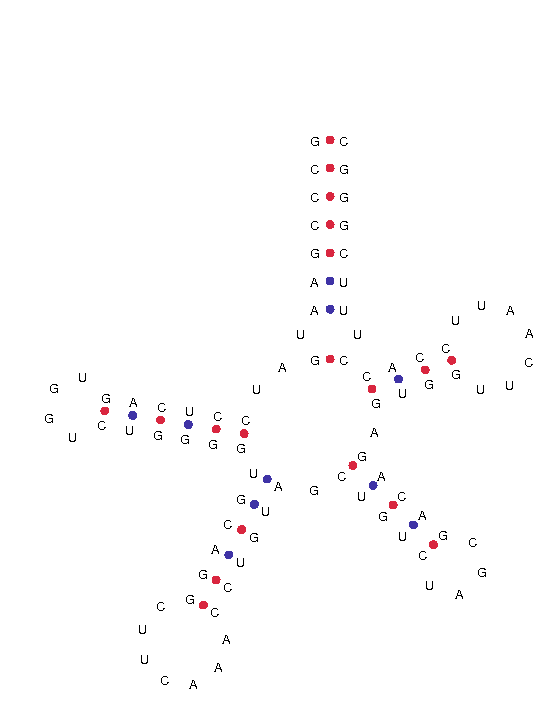{kind=link}
Selenoprofiles és una eina basada en homología que permet escanejar genomes en busca de selenoproteïnes que formen part de les famílies ja descrites. D’aquesta manera s’identifiquen tant selenoproteïnes reals com homòlegs en cisteïna.
Ha estat utilitzada per corroborar la feina feta amb la resta de programes, ja que executa de manera automatitzada alguns dels ja utilitzats: psitblastn, exonerate, genewise and SECISearch. D’aquesta manera combina diverses eines de comparació de genomes reduint substancialment el temps per fer l’scanning de genomes quan es busquen famílies de proteïnes.
4. Automatització
Per tal de fer possible l’anàlisi d’una gran quantitat de dades, hem creat alguns programes per automatitzar el procés. Podeu consultar aquí el codi font dels programes.
Up